Publications
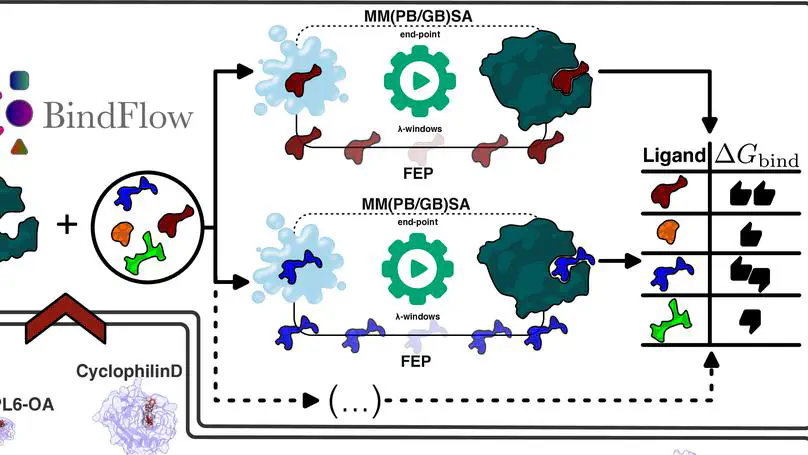
We present BindFlow, a Python-based software for automated absolute binding free energy (ABFE) calculations at the free energy perturbation (FEP) or at the molecular mechanics Poisson–Boltzmann/generalized Born surface area [MM(PB/GB)SA] level of theory. BindFlow is free, open-source, user-friendly, and easily customizable, runs on workstations or distributed computing platforms, and provides extensive documentation and tutorials. BindFlow uses GROMACS as a molecular dynamics engine and provides built-in support for the small-molecule force fields GAFF, OpenFF, and Espaloma, as well as support for user-provided custom force fields. We test BindFlow by computing affinities for 139 receptor–ligand pairs, involving eight different targets, including six soluble proteins, one membrane protein, and one nonprotein host–guest system. We find that the agreement of BindFlow predictions with experiments is overall similar to gold standards in the field. Interestingly, we find that MM(PB/GB)SA achieves correlations that, for some systems and force fields, approach those obtained with FEP while requiring only a fraction of the computational cost. This study establishes BindFlow as a validated and accessible tool for ABFE calculations.
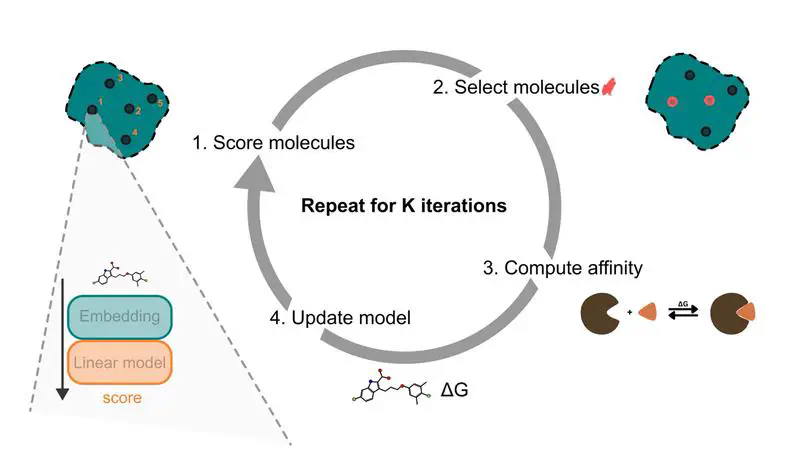

We present Moldrug, a computational tool for accelerating the hit-to-lead phase in structure-based drug design. Moldrug explores the chemical space using structural modifications suggested by the CReM library and by optimizing an adaptable fitness function with a genetic algorithm. Moldrug is complemented by Moldrug-Dashboard, a cross-platform and user-friendly graphical interface tailored for the analysis of Moldrug simulations. To illustrate Moldrug, we designed new potential inhibitors targeting the main protease (MPro) of SARS-CoV-2 by optimizing a consensus fitness function that balances binding affinity, drug-likeness, and synthetic accessibility. The designed molecules exhibited high chemical diversity. A subset of the designed molecules were ranked using MM/GBSA and alchemical binding free energy calculations, revealing predicted affinities as low as −10 kcal/mol. Moldrug is distributed as a Python package under the Apache 2.0 license. It offers pre-configured multi-parameter fitness functions for molecular design, while being highly adaptable for integrating functionalities from external software. Documentation and tutorials are available at https://moldrug.rtfd.io.